RNA-Seq-Data-Analysis
RNA-Seq-Data-Analysis Workflow
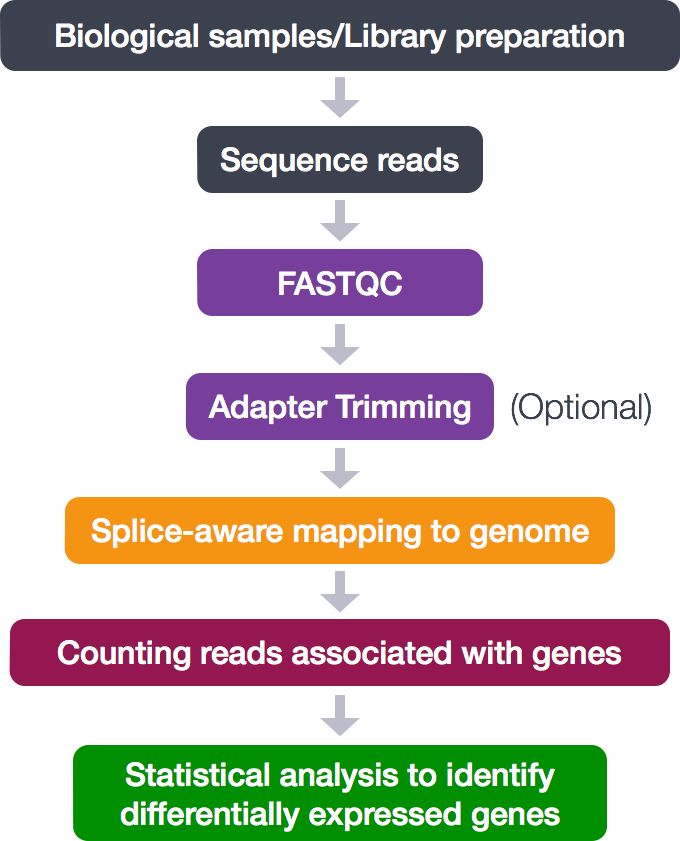
Analyzing RNA-seq data involves several steps to process raw sequencing reads into interpretable results. Here’s a generalized pipeline for RNA-seq data analysis:
Quality Control (QC):
Check the quality of raw sequencing reads using tools like FastQC or MultiQC. Trim adapters and low-quality bases using tools like Trimmomatic or Cutadapt.
Read Alignment:
Map the cleaned reads to a reference genome or transcriptome using alignment tools such as HISAT2, STAR, or Bowtie. Alternatively, align reads to transcriptome alone for certain analyses using tools like Salmon or Kallisto.
Quantification:
Estimate gene or transcript abundances from aligned reads. This step generates count data, which represents the expression level of each gene/transcript. Tools for quantification include featureCounts, HTSeq, Salmon, or Kallisto. Normalization:
Normalize the count data to account for sequencing depth and other technical biases. Common methods include TPM (Transcripts Per Million) or FPKM (Fragments Per Kilobase Million). Differential Expression Analysis:
Identify genes/transcripts that are differentially expressed between conditions (e.g., treatment vs. control). Popular tools for this step include DESeq2, edgeR, limma-voom, or Sleuth.
Functional Analysis:
Perform gene ontology (GO) analysis, pathway analysis, or other functional enrichment analyses to interpret the biological significance of differentially expressed genes. Tools like DAVID, Enrichr, or clusterProfiler are commonly used for functional analysis.
Visualization:
Visualize the results using various plots such as volcano plots, MA plots, heatmaps, or pathway enrichment plots. Tools like R (ggplot2, heatmap.2) or Python (matplotlib, seaborn) can be used for visualization. Validation:
Validate the findings using techniques such as qRT-PCR or Western blotting.
Annotation:
Annotate the differentially expressed genes/transcripts with relevant biological information using databases like NCBI, Ensembl, or UniProt.
Integration:
Integrate RNA-seq data with other omics data (e.g., ChIP-seq, ATAC-seq, proteomics) for a comprehensive understanding of biological processes.